from katlas.data import Data
from katlas.pssm import get_pssm_seq_labels,flatten_pssm
from kplot.hierarchical import get_hclusterhierarchical
Plot hierarchical clustering for PSSMs
Setup
pssms=Data.get_pspa_scale()distance = get_1d_js(pssms.head(20))100%|██████████| 20/20 [00:00<00:00, 360.94it/s]distance = get_1d_js_parallel(pssms.head(20))100%|██████████| 190/190 [00:00<00:00, 5548.41it/s]Pipeline of clustering
# pssms=Data.get_pspa_scale()
# Z = get_Z(pssms,func_flat=js_divergence_flat)
# count_dict = pssms.index.value_counts()
# labels= get_pssm_seq_labels(pssms,count_dict)
# plot_dendrogram(Z,dense=8,labels=labels,thr=0.125)Or directly use get_hcluster(include calculating Z and plot dendrogram)
count_dict = pssms.index.value_counts()
labels= get_pssm_seq_labels(pssms,count_dict)
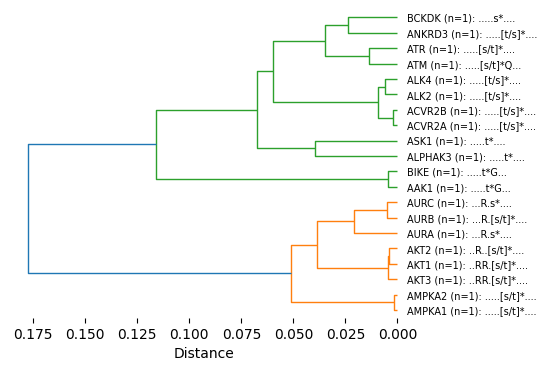
get_hcluster(pssms.head(20),func_flat=js_divergence_flat,labels=labels[:20],thr=0.125,dense=5)100%|██████████| 190/190 [00:00<00:00, 5515.69it/s]kinase
AAK1 2
ACVR2A 2
ACVR2B 2
AKT1 1
AKT2 1
AKT3 1
ALK2 2
ALK4 2
ALPHAK3 2
AMPKA1 1
AMPKA2 1
ANKRD3 2
ASK1 2
ATM 2
ATR 2
AURA 1
AURB 1
AURC 1
BCKDK 2
BIKE 2
dtype: int32