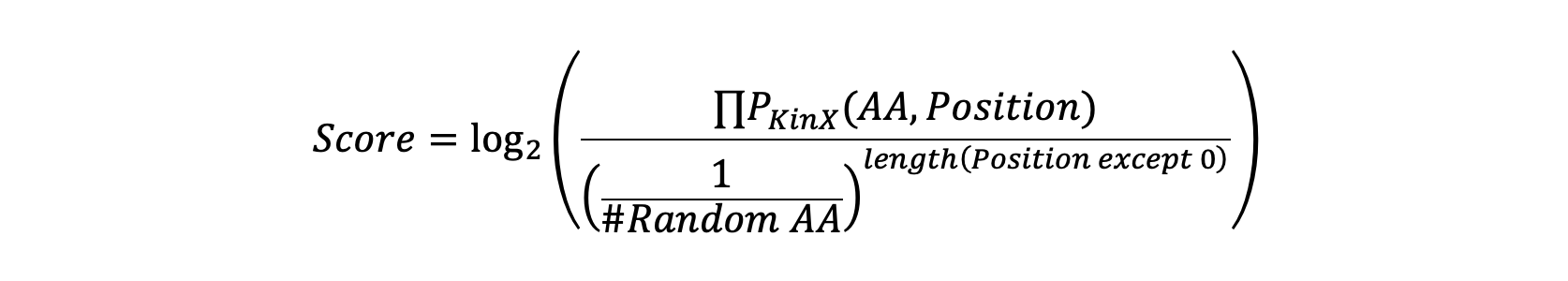
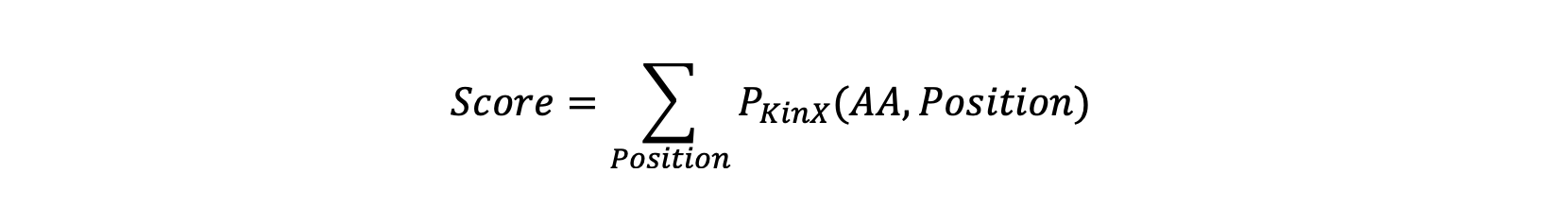multiply (values, kinase, num_dict={'SYK': 18, 'PTK2': 18, 'ZAP70': 18,
'ERBB2': 18, 'CSK': 18, 'FGFR4': 18, 'EGFR': 18, 'ERBB4': 18,
'EPHA8': 18, 'EPHA7': 18, 'EPHA5': 18, 'EPHA2': 18, 'EPHB2':
18, 'EPHB1': 18, 'EPHB3': 18, 'EPHB4': 18, 'EPHA4': 18,
'EPHA3': 18, 'EPHA6': 18, 'FRK': 18, 'EPHA1': 18, 'TEC': 18,
'BTK': 18, 'ITK': 18, 'BMX': 18, 'TXK': 16, 'ABL2': 18, 'ABL1':
18, 'SRMS': 18, 'PTK2B': 18, 'FER': 18, 'MERTK': 18, 'AXL': 18,
'FES': 18, 'PTK6': 18, 'YES1': 18, 'FGR': 18, 'SRC': 18, 'FYN':
18, 'LCK': 18, 'BLK': 18, 'LYN': 18, 'HCK': 18, 'PDGFRB': 18,
'PDGFRA': 18, 'FLT3': 18, 'TYRO3': 18, 'ROS1': 18, 'TEK': 18,
'LTK': 18, 'ALK': 18, 'MUSK': 18, 'KIT': 18, 'CSF1R': 18,
'MET': 18, 'KDR': 18, 'RET': 18, 'MST1R': 16, 'JAK3': 16,
'FLT1': 16, 'MATK': 18, 'FGFR3': 18, 'FGFR2': 18, 'FGFR1': 18,
'FLT4': 18, 'INSR': 18, 'IGF1R': 18, 'INSRR': 16, 'NTRK3': 18,
'NTRK1': 18, 'NTRK2': 18, 'TNK1': 18, 'TNK2': 18, 'DDR2': 18,
'DDR1': 18, 'TYK2': 18, 'JAK2': 18, 'JAK1': 18, 'TNNI3K_TYR':
18, 'NEK10_TYR': 16, 'PINK1_TYR': 16, 'MAP2K7_TYR': 16,
'PKMYT1_TYR': 16, 'TESK1_TYR': 16, 'LIMK1_TYR': 16,
'LIMK2_TYR': 16, 'WEE1_TYR': 18, 'MAP2K6_TYR': 16,
'MAP2K4_TYR': 16, 'PDHK1_TYR': 16, 'BMPR2_TYR': 16,
'PDHK4_TYR': 16, 'PDHK3_TYR': 16, 'AAK1': 17, 'ACVR2A': 17,
'ACVR2B': 17, 'AKT1': 17, 'AKT2': 17, 'AKT3': 17, 'ALK2': 17,
'ALK4': 17, 'ALPHAK3': 17, 'AMPKA1': 17, 'AMPKA2': 17,
'ANKRD3': 17, 'ATM': 17, 'ATR': 17, 'AURA': 17, 'AURB': 17,
'AURC': 17, 'GRK2': 17, 'GRK3': 17, 'BCKDK': 17, 'BIKE': 17,
'BMPR1A': 17, 'BMPR1B': 17, 'BMPR2': 17, 'BRAF': 17, 'BRSK1':
17, 'BRSK2': 17, 'BUB1': 17, 'CAMK1A': 17, 'CAMK1B': 17,
'CAMK1D': 17, 'CAMK1G': 17, 'CAMK2A': 17, 'CAMK2B': 17,
'CAMK2D': 17, 'CAMK2G': 17, 'CAMK4': 17, 'CAMKK1': 17,
'CAMKK2': 17, 'CAMLCK': 17, 'CDK1': 17, 'CDC7': 17, 'CDK10':
17, 'CDK19': 17, 'CDK2': 17, 'CDK3': 17, 'CDK4': 17, 'CDK5':
17, 'CDK6': 17, 'CDK7': 17, 'CDK8': 17, 'CDK9': 17, 'CDKL1':
17, 'CDKL5': 17, 'CHAK1': 17, 'CHAK2': 17, 'CDK13': 17, 'CHK1':
17, 'CHK2': 17, 'CK1A': 17, 'CK1A2': 17, 'CK1D': 17, 'CK1E':
17, 'CK1G1': 17, 'CK1G2': 17, 'CK1G3': 17, 'CK2A1': 17,
'CK2A2': 17, 'CLK1': 17, 'CLK2': 17, 'CLK3': 17, 'CLK4': 17,
'COT': 17, 'CRIK': 17, 'CDK12': 17, 'DAPK1': 17, 'DAPK2': 17,
'DAPK3': 17, 'DCAMKL1': 17, 'DCAMKL2': 17, 'DLK': 17, 'DMPK1':
17, 'DNAPK': 17, 'DRAK1': 17, 'DYRK1A': 17, 'DYRK1B': 17,
'DYRK2': 17, 'DYRK3': 17, 'DYRK4': 17, 'ERK1': 17, 'ERK2': 17,
'ERK5': 17, 'ERK7': 17, 'MTOR': 17, 'GAK': 17, 'GCK': 17,
'GCN2': 17, 'GRK4': 17, 'GRK5': 17, 'GRK6': 17, 'GRK7': 17,
'GSK3A': 17, 'GSK3B': 17, 'HASPIN': 17, 'HGK': 17, 'HIPK1': 17,
'HIPK2': 17, 'HIPK3': 17, 'HIPK4': 17, 'HPK1': 17, 'HRI': 17,
'HUNK': 17, 'ICK': 17, 'IKKA': 17, 'IKKB': 17, 'IKKE': 17,
'IRAK1': 17, 'IRAK4': 17, 'IRE1': 17, 'IRE2': 17, 'JNK1': 17,
'JNK2': 17, 'JNK3': 17, 'KHS1': 17, 'KHS2': 17, 'KIS': 17,
'LATS1': 17, 'LATS2': 17, 'LKB1': 17, 'LOK': 17, 'LRRK2': 17,
'MAK': 17, 'MEK1': 17, 'MEK2': 17, 'MEK5': 17, 'MEKK1': 17,
'YSK4': 17, 'MEKK2': 17, 'MEKK3': 17, 'ASK1': 17, 'MEKK6': 17,
'MAP3K15': 17, 'MAPKAPK2': 17, 'MAPKAPK3': 17, 'MAPKAPK5': 17,
'MARK1': 17, 'MARK2': 17, 'MARK3': 17, 'MARK4': 17, 'MASTL':
17, 'MELK': 17, 'MINK': 17, 'MLK1': 17, 'MLK2': 17, 'MLK3': 17,
'MLK4': 17, 'MNK1': 17, 'MNK2': 17, 'MOK': 17, 'MOS': 17,
'MPSK1': 17, 'MRCKA': 17, 'MRCKB': 17, 'MSK1': 17, 'MSK2': 17,
'SRPK3': 17, 'MST1': 17, 'MST2': 17, 'MST3': 17, 'MST4': 17,
'MYO3A': 17, 'MYO3B': 17, 'NDR1': 17, 'NDR2': 17, 'NEK1': 17,
'NEK11': 17, 'NEK2': 17, 'NEK3': 17, 'NEK4': 17, 'NEK5': 17,
'NEK6': 17, 'NEK7': 17, 'NEK8': 17, 'NEK9': 17, 'NIK': 17,
'NIM1': 17, 'NLK': 17, 'NUAK1': 17, 'NUAK2': 17, 'OSR1': 17,
'P38A': 17, 'P38B': 17, 'P38D': 17, 'P38G': 17, 'P70S6K': 17,
'P70S6KB': 17, 'PAK1': 17, 'PAK2': 17, 'PAK3': 17, 'PAK4': 17,
'PAK5': 17, 'PAK6': 17, 'PASK': 17, 'PBK': 17, 'CDK16': 17,
'CDK17': 17, 'CDK18': 17, 'PDHK1': 16, 'PDHK4': 16, 'PDK1': 17,
'PERK': 17, 'CDK14': 17, 'PHKG1': 17, 'PHKG2': 17, 'PIM1': 17,
'PIM2': 17, 'PIM3': 17, 'PINK1': 17, 'PKACA': 17, 'PKACB': 17,
'PKACG': 17, 'PKCA': 17, 'PKCB': 17, 'PKCD': 17, 'PKCE': 17,
'PKCG': 17, 'PKCH': 17, 'PKCI': 17, 'PKCT': 17, 'PKCZ': 17,
'PRKD1': 17, 'PRKD2': 17, 'PRKD3': 17, 'PKG1': 17, 'PKG2': 17,
'PKN1': 17, 'PKN2': 17, 'PKN3': 17, 'PKR': 17, 'PLK1': 17,
'PLK2': 17, 'PLK3': 17, 'PLK4': 17, 'PRKX': 17, 'PRP4': 17,
'PRPK': 17, 'QIK': 17, 'QSK': 17, 'RAF1': 17, 'GRK1': 17,
'RIPK1': 17, 'RIPK2': 17, 'RIPK3': 17, 'ROCK1': 17, 'ROCK2':
17, 'P90RSK': 17, 'RSK2': 17, 'RSK3': 17, 'RSK4': 17, 'SBK':
17, 'MYLK4': 17, 'SGK1': 17, 'SGK3': 17, 'DSTYK': 17, 'SIK':
17, 'SKMLCK': 17, 'SLK': 17, 'SMG1': 17, 'SMMLCK': 17, 'SNRK':
17, 'SRPK1': 17, 'SRPK2': 17, 'SSTK': 17, 'STK33': 17, 'STLK3':
17, 'TAK1': 17, 'TAO1': 17, 'TAO2': 17, 'TAO3': 17, 'TBK1': 17,
'TGFBR1': 17, 'TGFBR2': 17, 'TLK1': 17, 'TLK2': 17, 'TNIK': 17,
'TSSK1': 17, 'TSSK2': 17, 'TTBK1': 17, 'TTBK2': 17, 'TTK': 17,
'ULK1': 17, 'ULK2': 17, 'VRK1': 17, 'VRK2': 17, 'WNK1': 17,
'WNK3': 17, 'WNK4': 17, 'YANK2': 17, 'YANK3': 17, 'YSK1': 17,
'ZAK': 17, 'EEF2K': 17, 'FAM20C': 17})